
For the majority of the summer (save for my two week adventure), I have been continuing my research with M. monoensis in the lab. Much of our prior research focused on identifying a DNA barcode, a region that can be utilized to identify an unknown specimen. So recently, we established the ndhF-rpl32 region as a DNA barcode to differentiate between M. monoensis and its relatives: a SNP (single-nucleotide polymorphism) exists within the region. Mentzelia monoensis has the “G” allele and others (such as M. montana and M. albicaulis) have the “T” allele.
With the barcode, then, identification becomes less difficult–you just need to examine the specimen’s sequenced DNA. Sequencing, however, is expensive. Often, we pay upwards of $6 for each sample’s sequence. Individually, this figure seems insignificant (I mean we pay $50 for our gel combs, right?). But given 100 or so samples, that $6 easily becomes $600. This can hamper small labs with limited funds and, in turn, our efforts to establish the true distribution of M. monoensis. Thus, using our understanding of the SNP, we decided to investigate more cost-effective methods of identification. We found one method, known as SNP haplotyping, particularly promising. This approach primarily utilizes allele-specific PCR and gel electrophoresis, which are both cost-effective procedures.
In order to begin our work with SNP haplotyping, we first needed to design primers–they would be the key to the project’s success. The process itself requires three primers total: a common reverse primer and two forward allele-specific primers. The common primer didn’t require much alteration; we just made sure it worked well with our sequence. However, the allele-specific primers required a few additions. Specifically, we added destabilizing mismatches within five bases of the 3’ ends, which facilitated primer specificity and, thus, identification. Additionally, we had to attach 5’ tails to our primers. The lengths of said tails differed by about 10 base pairs, which allowed us to distinguish the different species on a gel. This was definitely one of the more difficult aspects of the project, especially as Dr. Brokaw and I both had limited experience with primer design. We suddenly had to consider GC content (for the Tm), primer dimers (when the primers bind to each other), and hairpin loops (when the DNA folds back on itself) among other things. Though with the help of Dr. Huddleston (she’s awesome!), we were able to design a common primer and 7 (yes, 7) potential primer pairs. Why 7? Well, often (as I now understand) primers tend to be a mixed bag. Sometimes they work and sometimes…not so much.
Once we received our primers, we began testing them using DNA from a few samples of M. montana, M. monoensis, and M. albicaulis. We ran many PCRs and many gels. With those PCRs and gels came many failures and disappointments. We soon discovered that most, if not all, of our primers behaved inconsistently. Some primer pairs didn’t even amplify at all! And don’t even get me started about our negative controls. In fact, until about two weeks ago, we weren’t quite sure if this project would have a “successful” result. But, lo and behold, we found hope in primer pair 3 after testing a few of the primer pairs again. The M. monoensis samples (whose AS primers had shorter tails) traveled noticeably further than the M. albicaulis or M. montana samples. So, what was so special about primer pair 3? Well…we’re not quite sure. But primer pair 3, interestingly enough, was the only pair to have 2 strong mismatches (ie a purine replacing a pyrimidine, etc.). All the other pairs had combinations of weak (ie a pyrimidine replacing a pyrimidine, etc.) and strong mismatches. In the future, it would be interesting to design primer pairs with only strong mismatches–perhaps more of our primers would work? But quite honestly, I am thrilled that primer pair 3 even cooperated at all.
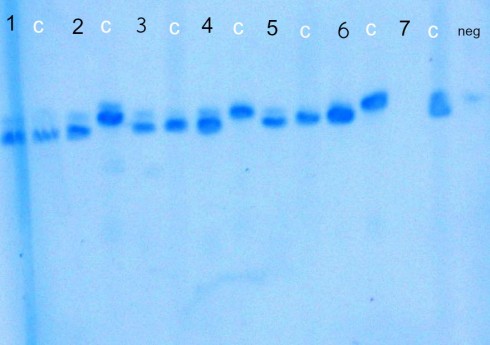
Primer Pair 3
Order (from left to right): 1. M. monoensis 547 , M. monoensis control, 2. M. monoensis 554, M. montana control, 3. M. monoensis 556, M. monoensis control, 4. M. monoensis 557, M. montana control, 5. M. monoensis 558, M. monoensis control, 6. M. monoensis 559, M. montana control, 7. M. monoensis 560 (didn’t amplify), M. monoensis control, negative control (with faint band)
Along the way, we made some important discoveries about some of our materials and methods. Early on we made the switch from using TAE buffer to TBE buffer, as some suggested TBE worked well with smaller products (ours are around 200 base pairs) and produces better band resolution. Next, we transitioned to MetaPhor agarose because it can distinguish small PCR products and improve the clarity of the gel. Despite the extra expense, it seems to be working better than the standard agarose (for our purposes at least). Our most important change, by far, was our transition from SYBR-Green to ethidium bromide (SYBR-Green is people!). Not only that, but now we stain our gels after they’ve run (another change: gels now run in the refrigerator). It is a bit scary handling a carcinogen, but ethidium bromide has facilitated the project. Also, we have been tinkering with the voltage and length of our gels. Initially, we tried smaller (about 40 V) voltages, hoping this would allow us to best distinguish between small distances. However, we now realize that “if the voltage is too low, then the mobility is reduced and band broadening will occur due to diffusion.” So now we’re trying to run our gels at higher voltages (90-120 V) for longer increments of time (3-4 hours). We haven’t quite exactly pinpointed these conditions, but as we continue on with this project we hope to discover the “ideal” setting.
I’m so glad I have been able to continue my research with Dr. Brokaw this summer. I feel like I’m finally getting comfortable around the lab–with the equipment, methods, and the people even. It was nice to be able to focus solely on research, which gave me a more realistic picture of grad school and the research field as a whole. This summer I encountered difficulties and made mistakes, but I also found successes and became more confident in my own abilities. It seems bittersweet to think that my summer work is coming to a close, but I am definitely excited to move forward with this project during the semester (preview of coming attractions: we will use primer pair 3 to identify many samples)!